Short Notes Index
G-Proteins
Cytochrome P450
Formation & fate of ammonia in the body
Why is ammonia toxic to the body?
Biologically important products formed from phenylalanine & tyrosine.
Inborn errors, biochemical abnormalities, and manifestations associated with the metabolism of phenylalanine & tyrosine.
Von-Gierke's disease
Tubular Maximum for Glucose (TmG)
Protein Synthesis Process
DNA Repair Mechanism
G-Proteins
✅ Definition in one line for exams:
“G-proteins are guanine nucleotide-binding proteins that act as molecular switches in signal transduction, coupling receptors to intracellular effectors.”
A G-protein (Guanine nucleotide-binding protein) is a molecular switch inside cells that helps transmit signals from cell surface receptors to intracellular targets.
They are called G-proteins because they bind guanosine triphosphate (GTP) and guanosine diphosphate (GDP).
G-proteins are involved in signal transduction — converting an extracellular signal (like a hormone, neurotransmitter, or light) into a cellular response.
🔎 Key Features
1. Location → Found on the inner surface of the plasma membrane, linked to G-protein-coupled receptors (GPCRs).
2. Subunits → Usually exist as a heterotrimer with α, β, and γ subunits.
o α-subunit binds GDP/GTP and has GTPase activity.
o βγ complex also participates in signaling.
3. Switch Mechanism:
o Inactive state → bound to GDP.
o Active state → when the receptor stimulates, GDP is replaced by GTP → the G-protein activates downstream effectors.
o Later, α-subunit hydrolyzes GTP → GDP (turns itself off).
⚙️ Functions
Activate or inhibit enzymes like adenylate cyclase or phospholipase C.
Regulate ion channels.
Control production of second messengers (e.g., cAMP, IP₃, DAG).
Clinical Significance
Many hormones (e.g., adrenaline, glucagon, ACTH) act via G-proteins.
Diseases can result from G-protein malfunction (e.g., cholera toxin locks G-protein in active state → ↑cAMP → diarrhea).
✅ Definition (short form):
“Cytochrome P450 is a superfamily of heme-containing monooxygenase enzymes that catalyze drug metabolism and biosynthesis of steroids, located mainly in the liver.”
Cytochrome P450 (CYP450)
Cytochrome P450 (CYP450) is a large family of heme-containing enzymes that act as monooxygenases.
Location: They are mainly found in the smooth endoplasmic reticulum and mitochondria of cells.
Why "P450": They are named “P450” because they absorb light at 450 nm when bound to carbon monoxide.
🔎Key Functions
1. Drug Metabolism (Phase I reactions)
o Oxidation, reduction, and hydroxylation of drugs.
o Converts lipid-soluble drugs into more water-soluble metabolites (often preparing them for Phase II conjugation).
2. Endogenous Metabolism
o Steroid hormone synthesis (cortisol, estrogen, testosterone).
o Vitamin D metabolism.
o Metabolism of fatty acids, bile acids, and prostaglandins.
Reaction (General)
RH+O2+NADPH+H+→ROH+H2O+NADP+RH + O_2 + NADPH + H^+ → ROH + H_2O + NADP^+
RH = substrate (drug or compound)
Adds an -OH group (hydroxylation)
📍 Location
Liver (major site) → drug detoxification.
Also in the intestine, lungs, kidneys, and adrenal glands.
Clinical Relevance
Drug interactions:
Some drugs induce CYP450 → increase metabolism of other drugs (e.g., rifampicin, phenytoin).
Some drugs inhibit CYP450 → slow metabolism, increase toxicity risk (e.g., erythromycin, ketoconazole).
Genetic polymorphisms:
Different CYP450 isoenzymes vary among individuals, → explains why some patients metabolize drugs faster/slower.
Toxins/Carcinogens:
Can activate pro-carcinogens (e.g., benzopyrene from cigarette smoke).
Formation & fate of ammonia in the body
1. Formation of Ammonia (Sources in the Body)
Ammonia is produced continuously as a byproduct of amino acid and nucleotide metabolism.
Main sources:
Oxidative deamination
Amino acids → α-keto acids + NH₃
Enzyme: Glutamate dehydrogenase (in mitochondria).
Non-oxidative deamination
Serine, threonine, and cysteine can release NH₃ directly.
Hydrolytic deamination
Glutamine → glutamate + NH₃ (enzyme: glutaminase).
Asparagine → aspartate + NH₃ (enzyme: asparaginase).
Purine & pyrimidine catabolism
AMP, GMP → NH₃ released.
Bacterial action in the intestine
Gut flora deaminates urea and amino acids → NH₃ absorbed into portal circulation.
2. Fate of Ammonia
Since free ammonia is toxic, the body quickly detoxifies it:
(A) Transport in blood (non-toxic forms)
Glutamine pathway: NH₃ + glutamate → glutamine (enzyme: glutamine synthetase).
Alanine pathway (muscle → liver): Pyruvate + NH₃ → alanine (glucose-alanine cycle).
(B) Detoxification in the liver
Urea cycle (main disposal, occurs in the liver):
NH₃ + CO₂ → carbamoyl phosphate → urea.
Urea is transported via blood → excreted in urine.
Glutamine formation (temporary storage form of ammonia, esp. in the brain).
(C) Excretion routes
Urea (~80–90% of nitrogen excretion).
Ammonium salts (~2–5%, important for acid-base regulation in the kidney).
Creatinine, uric acid (minor pathways).
Why is ammonia toxic to the body?
3. Why Ammonia is Highly Toxic
Free ammonia, even in small amounts, disrupts CNS function.
Mechanisms of toxicity:
Neurotoxicity
NH₃ readily crosses the blood-brain barrier.
In the brain: NH₃ + glutamate → glutamine.
This depletes glutamate (a major excitatory neurotransmitter) → impaired neuronal function.
Excess glutamine increases osmotic pressure → cerebral edema.
Energy depletion
Detoxification of NH₃ consumes α-ketoglutarate (in the TCA cycle).
Depletion of α-KG → inhibition of TCA cycle → ↓ ATP → neuronal dysfunction.
Acid-base imbalance
Excess NH₃ alters H⁺ buffering in the kidney → metabolic disturbances.
4. Clinical Aspect
Hyperammonemia is seen in:
Liver failure (cirrhosis, viral hepatitis).
Inborn errors of urea cycle enzymes.
Symptoms: Irritability, vomiting, tremors, cerebral edema, convulsions, coma, death.
✅Key Points
Formation → Amino acid catabolism (oxidative, non-oxidative, hydrolytic), purine/pyrimidine breakdown, intestinal bacteria.
Fate→ Transport as glutamine/alanine → liver detox → urea cycle → excretion as urea/NH₄⁺ salts.
Toxicity → Neurotoxic (glutamate depletion, cerebral edema), inhibits the TCA cycle (ATP deficiency), acid-base imbalance.
Biologically important products formed from phenylalanine & tyrosine.
1. From Phenylalanine
Tyrosine itself → formed by hydroxylation of phenylalanine (enzyme: Phenylalanine hydroxylase, requires BH₄).
⚡ Thus, most of phenylalanine’s importance is via conversion to tyrosine.
2. From Tyrosine
(A) Catecholamines (Neurotransmitters & Hormones)
Pathway:
Tyrosine → DOPA → Dopamine → Noradrenaline → AdrenalineFunctions:
Dopamine: neurotransmitter (movement, reward pathway).
Noradrenaline: sympathetic neurotransmitter.
Adrenaline: stress hormone (fight-or-flight).
(B) Thyroid Hormones
Tyrosine residues in thyroglobulin undergo iodination → T₃ (triiodothyronine) & T₄ (thyroxine).
Functions: Regulate basal metabolic rate, growth, and development.
(C) Melanin (Pigment)
Tyrosine → DOPA → DOPA quinone → Melanin (enzyme: Tyrosinase).
Functions: Skin, hair, eye pigmentation; UV protection.
Clinical: Deficiency of tyrosinase → Albinism.
(D) Fumarate & Acetoacetate (Energy metabolism)
Tyrosine is both glucogenic (fumarate) and ketogenic (acetoacetate).
Important in energy pathways.
(E) Other Minor Products
Enkephalins: opioid peptides derived partly from tyrosine.
Tyramine: vasoactive amine (from tyrosine decarboxylation).
Inborn errors, biochemical abnormalities, and manifestations associated with the metabolism of phenylalanine & tyrosine.
1. Phenylketonuria (PKU)
Defect: Phenylalanine hydroxylase deficiency (or BH₄ deficiency).
Biochemical abnormality: ↑ Phenylalanine, ↓ Tyrosine, abnormal phenylketones in urine.
Clinical features: Severe intellectual disability, seizures, “mousy odour” urine, hypopigmentation (↓ melanin).
2. Alkaptonuria
Defect: Homogentisate oxidase deficiency.
Biochemical abnormality: ↑ Homogentisic acid in blood & urine.
Clinical features: Black urine on standing, ochronosis (bluish-black pigmentation of cartilage, sclera), arthritis.
3. Tyrosinemia Type I (Hepatorenal Tyrosinemia)
Defect: Fumarylacetoacetate hydrolase deficiency.
Biochemical abnormality: Accumulation of fumarylacetoacetate & succinyl acetone.
Clinical features: Liver failure, renal tubular dysfunction, rickets, cabbage-like odour, fatal in infancy if untreated.
4. Tyrosinemia Type II (Richner–Hanhart Syndrome)
Defect: Tyrosine aminotransferase deficiency.
Biochemical abnormality: ↑ Plasma tyrosine.
Clinical features: Keratitis, painful skin lesions (palms/soles), intellectual disability.
5. Tyrosinemia Type III (Rare)
Defect: p-Hydroxyphenylpyruvate dioxygenase deficiency.
Biochemical abnormality: ↑ Tyrosine & p-hydroxyphenylpyruvate.
Clinical features: Intellectual disability, seizures, ataxia.
6. Albinism
Defect: Tyrosinase deficiency (or other steps in melanin synthesis).
Biochemical abnormality: No melanin formation from tyrosine.
Clinical features: Hypopigmentation of skin, hair, and eyes; photophobia; ↑ risk of skin cancers.
7. Disorders of Catecholamine Synthesis
Dopamine β-hydroxylase deficiency:
Biochemical abnormality: ↓ Noradrenaline & adrenaline.
Clinical: Orthostatic hypotension, ptosis, hypoglycemia.
Parkinson’s disease (not congenital, but relevant):
Loss of dopaminergic neurons → tremors, rigidity, bradykinesia.
8. Thyroid Hormone Disorders
Defect: Defects in tyrosine iodination & coupling (congenital hypothyroidism).
Biochemical abnormality: ↓ T₃ & T₄.
Clinical features: Cretinism (mental retardation, dwarfism), goiter.
Von-Gierke's disease
4. Clinical Featuresa
Usually appear in infancy when fasting tolerance is tested.
Severe fasting hypoglycemia → irritability, seizures.
Hepatomegaly (due to glycogen accumulation).
Doll-like face with fat cheeks.
Protuberant abdomen (enlarged liver + kidneys).
Growth retardation, delayed puberty.
Type Ib: recurrent infections & neutropenia (due to defect in neutrophil function).
5. Investigations
Blood glucose: very low (especially during fasting).
↑ Lactic acid, ↑ uric acid, ↑ and triglycerides.
Liver biopsy: massive glycogen deposition.
Genetic testing confirms enzyme defect.
6. Treatment
Frequent feeding with cornstarch (slow-release glucose → prevents fasting hypoglycemia.
Avoid galactose & fructose (they enter the pathway as G6P, worsening the metabolic block).
Allopurinol for hyperuricemia.
Granulocyte colony-stimulating factor (G-CSF) in Type Ib (for neutropenia).
7. Clinical Importance (as per MCI/DCI emphasis)
Teaches integration of biochemistry with clinical signs (classic example in exams).
Links glycogen metabolism with hypoglycemia, metabolic acidosis, and hepatomegaly.
Provides rationale for dietary treatment in metabolic diseases.
1. Introduction
First described by Edgar von Gierke.
It is the most common glycogen storage disease (GSD-I).
An autosomal recessive disorder affecting glycogen metabolism.
2. Enzyme Deficiency
Glucose-6-phosphatase deficiency (Type Ia)
Enzyme normally converts glucose-6-phosphate → free glucose, the final step of glycogenolysis and gluconeogenesis.
Glucose-6-phosphate translocase deficiency (Type Ib).
⚡ Key Concept: Because of this block, glycogen cannot be broken down to free glucose → severe fasting hypoglycemia.
3. Biochemical Basis / Pathophysiology
Excess glycogen accumulation in the liver and kidneys (since glucose cannot be released).
Hypoglycemia (since glycogenolysis & gluconeogenesis are both blocked).
Lactic acidosis (excess G6P shunted into glycolysis → pyruvate → lactate).
Hyperuricemia (competition between lactate and uric acid for renal excretion).
Hyperlipidemia (↑ acetyl-CoA, ↑ VLDL synthesis).
✅ Key Points:
AR disorder, deficiency of Glucose-6-phosphatase / G6P translocase.
Liver & kidney glycogen accumulation.
Hypoglycemia + 4 H’s (↑ lactate, uric acid, lipids).
Clinical: hepatomegaly, doll face, seizures, growth delay.
Treatment: frequent cornstarch, avoid fructose/galactose.
Tubular Maximum for Glucose (TmG)
Tubular Maximum for Glucose (TmG) is the maximum amount of glucose that the renal tubules can reabsorb per minute. Beyond this threshold, excess glucose remains in the filtrate and is excreted in urine.
Mechanism
Glucose is filtered freely at the glomerulus.
Reabsorbed in the proximal convoluted tubule via sodium-glucose transporters (SGLT2).
Once transporters are saturated, excess glucose is lost in urine.


Clinical Significance
📊 Normal Value
TmG in adults: Approximately 375 mg/min
Renal threshold for glucose: Around 180–200 mg/dL in blood
🧬 DNA Repair Mechanism
DNA repair is a vital cellular process that corrects damage to the DNA molecules to maintain genetic stability and prevent mutations. Damage can occur due to radiation, chemicals, replication errors, or oxidative stress.
🔧 Major Types of DNA Repair Mechanisms
1. Base Excision Repair (BER)
Fixes small, non-helix-distorting base lesions.
Involves the removal of the damaged base by DNA glycosylase and replacement by DNA polymerase.
2. Nucleotide Excision Repair (NER)
Removes bulky lesions like thymine dimers caused by UV radiation.
Involves the excision of a short DNA segment and resynthesis.
3. Mismatch Repair (MMR)
Corrects errors missed during DNA replication (e.g., base mismatches).
Defects in MMR are linked to hereditary nonpolyposis colorectal cancer.
4. Double-Strand Break Repair
Homologous Recombination (HR): Accurate repair using the sister chromatid as a template.
Non-Homologous End Joining (NHEJ): Direct ligation of broken ends; error-prone.
⚕️ Clinical Relevance
Cancer: Defective repair mechanisms can lead to genomic instability and tumor formation.
Genetic Disorders: Xeroderma pigmentosum (defective NER), Lynch syndrome (defective MMR).
Therapeutics: PARP inhibitors target defective HR in cancer cells (e.g., BRCA mutations).


🧬 Protein Synthesis Process
Protein synthesis is the biological process by which cells build proteins based on genetic instructions encoded in DNA. It occurs in two main stages: Transcription and Translation.
2. Translation (mRNA → Protein)
Location: Cytoplasm (at ribosomes)
Steps:
mRNA binds to a ribosome.
Transfer RNA (tRNA) brings amino acids to the ribosome.
Each tRNA has an anticodon that matches the mRNA codon.
The ribosome links amino acids via peptide bonds to form a polypeptide chain.
Process continues until a stop codon is reached.
The newly formed protein is released and undergoes folding/modification.
1. 🧾 Transcription (DNA → mRNA)
Location: Nucleus
Steps:
DNA unwinds and exposes the gene sequence.
RNA polymerase binds to the promoter region of DNA.
Complementary RNA nucleotides are assembled to form messenger RNA (mRNA).
mRNA detaches and exits the nucleus through nuclear pores.
🧠 Clinical Relevance
Antibiotics: Many target bacterial ribosomes to inhibit protein synthesis (e.g., tetracycline).
Genetic Disorders: Mutations in DNA can lead to faulty proteins (e.g., cystic fibrosis).
Cancer: Abnormal regulation of protein synthesis contributes to uncontrolled cell growth.

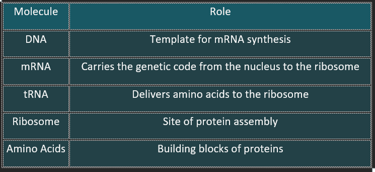
Key Molecules Involved
BLOG
Explore the medical biochemistry intricacies.
WRITE TO US
© 2024. All rights reserved.